В геноме человека содержится около 30 тысяч генов. Но повреждение даже одного из них может привести к страшным последствиям. Что уж говорить о целой хромосоме, которых всего-то 23 пары!
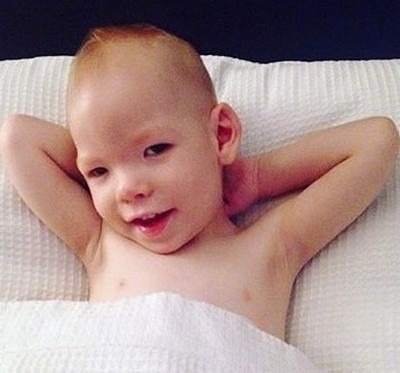
Этот синдром вызывается мутацией хромосомы 5 (5р-). Впервые описан французским педиатром Жеромом Лежёном в 1963 году и получил его имя. Кстати, именно Лежён нашел объяснению синдрому Дауна — лишняя хромосома в 21 паре.
Дети с такой аномалией, рождаясь, издают необычный крик, напоминающий мяуканье кошки. Но «кошачий плач», увы, самое безобидное проявление болезни. Оно вызывается изменением гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки). По сути синдром Лежёна — это глубокая умственная и физическая неполноценность.
У детей с синдромом кошачьего крика характерная внешность: лунообразное лицо, микроцефалия, антимонголоидный разрез глаз, косоглазие, высокое нёбо, плоская спинка носа. Ушные раковины деформированы и низко расположены. Кроме того, встречаются врождённые пороки сердца и некоторых других внутренних органов, косолапость, мышечная слабость.
Большинство больных умирают в первые годы жизни.
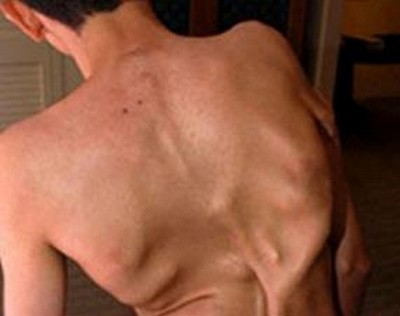 Каменная болезнь
Каменная болезньНаучное название - прогрессивная оссифицирующая фибродисплазия (ФОП). Это врожденная патология возникает из-за мутации гена ACVR1/ALK2 и встречается у одного из 2 миллионов жителей земли.
У страдающих ФОП травмы или воспалительные процессы, происходящие в мышцах, связках, сухожилиях и других соединительных тканях, приводят к кальцинированию и превращению этих тканей в кость. Фибродисплазию еще называют «болезнь второго скелета»: дефектный ген вызывает непрекращающийся рост костей (оссификатов) в тех местах, где их быть не должно.
Генетическая болезнь начинается в возрасте до 10 лет. У ребенка появляются бугры, чаще на спине. Врачи обычно подозревают онкологию, делается множество анализов и исследований, прежде чем диагноз будет поставлен. При этом любой укол или ушиб может вызвать рост оссификата.
Заболевание протекает с разной скоростью у разных больных. Грозит полной неподвижностью и ранней смертью.
В последнее время для больных ФОП появилась надежда: в 2006-м году ученым удалось обнаружить, какое именно генетическое отклонение приводит к образованию «второго скелета», а сейчас испытывают препарат, останавливающий рост патологической ткани.
 Прогерия
ПрогерияРедкий, но широко известный генетический дефект - преждевременное старение организма. Основными формами является детская прогерия – синдром Гетчинсона-Гилфорда и прогерия взрослых – синдром Вернера.
Прогерия взрослых (дефектный ген — WRN) проявляется старческими изменениями кожи и мышц, развитием катаракты, диабета, атеросклероза и других «стариковских» болезней. Проявляется в период полового созревания, встречается чаще всего у мужчин. После 20 лет у них седеют и выпадают волосы, руки и ноги становятся тонкими.
Прогерия детская характерна пропорциональной карликовостью, отсутствием подкожной клетчатки и ломкостью костей. Причина детской прогерии — мутации гена LMNA. Хотя детская прогерия может быть врождёенной, у большинства признаки проявляются обычно на втором-третьем году жизни.
Резко замедляется рост ребенка, кожа, особенно на лице и руках. Больной приобретает характерный внешний вид: большая голова, лобные бугры выступают над маленьким заостренным («птичьим») лицом с клювовидным носом, нижняя челюсть недоразвита. Атрофируются мышцы, дегенеративные процессы происходят в зубах, волосах, ногтях, костях и мышцах.
Лечения не существует, больные обычно умирают от атеросклероза или онкозаболеваний.
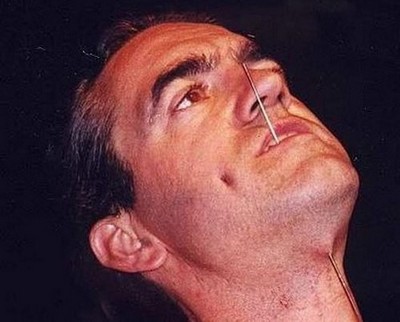 Нечувствительность к боли
Нечувствительность к болиЗаболевание называется «сенсорная полинейропатия». Поврежденный мутацией ген NGFB вызывает нечувствительность болевых рецепторов.
Больные не чувствуют боли, но способны ощущать вкус, прикосновение, давление и вибрацию. То есть нежное поглаживание они почувствуют, а вот сильный удар — нет!
Вдобавок к этому изменяется восприятие температуры, поэтому человек не может адекватно воспринимать холодное и горячее, что и приводит к ожогам и обморожениям.
У таких пациентов еще с детского возраста выявляется разрушение суставов по типу болезни Шарко. Это приводит к тому, что нарушается походка, человек не может выполнять сложные движения и т.д.
В отличие от других видов полиневропатий дети при такой мутации не отстают в умственном развитии. Кроме того, у них не развивается мышечная атрофия.
Лечение малоэффективно, обычно используют гормональные препараты.
Боль — это защитная реакция организма, сигнал о каких-то проблемах, поэтому полная потеря болевых ощущений — вовсе не благо. Больные все время рискуют навредить себе — прокусить язык, подвернуть или сломать ногу, порезаться и т. д.
На Востоке некоторые люди, родившиеся с нечувствительностью, зарабатывают себе на жизнь, устраивая уличные представления, где протыкают себе тело или причиняют другие травмы.
Особенность не чувствовать боли активно используется кинематографом: в 2012 году вышел испано-франко-португальский фильм «Нечувствительный», где рассказывалось о целой группе таких детей.
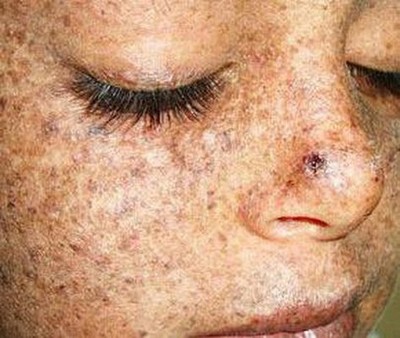 Пигментная ксеродерма
Пигментная ксеродермаДругие названия - ретикулярный прогрессирующий меланоз, прогрессирующий ретикулярный меланоз Пика. Это гиперчувствительность к солнечным лучам — ультрафиолету и радиации. Встречается примерно у четырех человек из миллиона. Возникает из-за мутации белков, ответственных за исправление повреждений ДНК, появляющихся при воздействии ультрафиолетового излучения.
Первые симптомы обычно возникают в возрасте до трех лет. Даже после нескольких минут пребывания на солнце у ребенка возникают серьезные ожоги. Постепенно открытые участки тела покрываются пятнами, веснушками, кожа начинает шелушиться.
Через несколько лет кожа становится пестрой — некоторые участки несут признаки атрофии, другие потемнели, видны сосудистые звездочки. Появляются бородавки, корки, трещины, язвы. Деформируются нос, уши, рот, глаза страдают от воспаления и слезотечения.
Больным приходится все время принимать препараты, снижающие светочувствительность, пользоваться мазями и кремами, удалять бородавчатые разрастания.
К подростковому возрасту образования на коже часто переходят в злокачественные. Две трети больных умирает до 15 лет.
Это генетическое заболевание очень «любят» кинематографисты. Персонажи, болеющие ксеродермой, присутствуют по меньшей мере в 10 фильмах.
